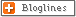Heart Structure and Blood Supply
It seems odd that the tissues making up the heart must have their own separate blood supply. You might think that the torrent of blood rushing through the heart every minute would more than adequately meet the needs of the organ. The walls of the heart, however, consist of layers of specialized muscle. These walls are quite thick—the wall of the left ventricle is often over 1 inch thick. Since the lining of the heart is watertight, the blood cannot seep through the layers of muscle to provide the nourishment essential to these constantly working masses. Blood is carried through the muscle layers that form the heart wall by means of the two coronary arteries. These two small vessels branch off the aorta just after it leaves the heart and curl back across the surface of the chambers, sending twigs through the walls (Fig. 4-1).
The coronary arteries are so named because of the supposed resemblance to a crown or “corona†of the little arteries as they encircle the heart. These arteries divide into smaller and smaller branches, like all blood vessels in the body, until they become so small that only one blood cell at a time can move through them. At this point the vessels are called capillaries. After the blood has passed through the capillaries, and the tissues have extracted the needed oxygen, it returns by way of veins, which become larger and larger until they, like all other veins in the body, empty into the right atrium. The veins from the wall of the heart, or coronary veins, empty into the right atrium through a structure called the coronary sinus.
The blood supply of the tissues in the wall of the heart is not very good; thousands of people die every year because of this curious fact. Most organs and tissues of the body have a “reserve†or collateral blood supply. Each finger, for instance, has two arteries, one on each side. These arteries are connected by many cross-channels, or collateral vessels. If the artery is cut on one side, the collateral or cross-connections from the artery on the other side would probably provide sufficient blood to maintain life in the tissues of the finger. The same “safety†feature is true in most of the major areas of the body. It is not true in the wall of the heart.
The coronary arteries tend to be end arteries, meaning that each branch follows its own course to some area of the heart muscle with relatively few connections to other branches nearby. If one of these coronary branches is plugged by hardening or by a blood clot, the muscle that depends on it for blood will die. A form of gangrene actually sets in. (Some people's coronary arteries have many more cross-connections than others. The more of these cross-connections an individual has, the less likely he or she is to die of coronary artery disease. In 10,000 or 20,000 years the process of evolution may result in a race with a good coronary blood supply by virtue of the early death of those without it.)
The names of the chief branches of the coronary arteries are important because they will be used repeatedly in this book. Learn them now; they're very simple.
There are two main coronary arteries leading out of the aorta—the right and left coronary arteries. After about an inch, the left coronary artery divides into two principal branches. The left anterior descending branch comes down the front of the heart, roughly along the septum between the two ventricles. The circumflex branch of the left coronary artery coils around the left side and back of the heart. The right coronary artery divides into a number of branches that course through the right chambers of the heart as well as through a large part of the left ventricle.

Note: There are four coronary arteries to remember:
The left main coronary artery (before it divides): LMCA.
The right coronary artery: RCA.
The left anterior descending branch of the left main coronary artery: LAD.
The circumflex branch of the left main coronary artery: LCA or LCirc.